

Measures of inflammation in atherosclerotic cardiovascular disease
Measures of inflammation in atherosclerotic cardiovascular disease
A deeper look into evidence in support of measuring hsCRP as means of assessing cardovascular risk

In Notes on notions I described how “In situations where it’s impossible to act as if one has full confidence in their strategy, the best thing one can do is to follow the totality of the evidence, all things considered, and draw conclusions that allow one to be pragmatic and trust the process.”
One of the best-established relationships in modern medicine is the causal link between apolipoprotein B (apoB) bearing particles and atherosclerotic cardiovascular disease (ASCVD).1 The “totality of evidence”, ranging from mechanistic research to case studies to epidemiological studies to randomized controlled trials to mendelian randomization and beyond suggests that apoB (or LDL-cholesterol or non-HDL-cholesterol) levels are not only associated, but causally contributing to the presence, development, and progression of atherosclerotic cardiovascular disease. Following the reasoning above, in light of the current evidence, maintaining high confidence in the validity of this theory wouldn’t seem ill-informed.
The details on how exactly apoB particles contribute to the initiation and development of ASCVD is a nuanced and debated subject, but on a macroscopic level apoB is seemingly conditionally necessary, but not sufficient for atherogenesis to occur.2 This has given rise, especially in the circles of cardiology and lipidology, to the mantra “the earlier, the deeper, the longer, the better”, referring to the expected benefits of reducing quantity- and time-exposure to apoB. For more: The Expected 30-Year Benefits of Early Versus Delayed Primary Prevention of Cardiovascular Disease by Lipid Lowering and There is urgent need to treat atherosclerotic cardiovascular disease risk earlier, more intensively, and with greater precision: A review of current practice and recommendations for improved effectiveness, as well as my previous writings here, and here.
Now, we need to acknowledge the limitations that the probabilistic conditioning described above introduces. Firstly, due to the seemingly stochastic nature by which apoB contributes to atherogenesis, as long as levels are above zero (which in practice they always are), there’s a theoretical non-zero probability that atherosclerosis will develop. Secondly, on the flip side, there’s always a small but non-zero probability that atherogenesis might not occur, or at least cause any issues, even in the presence of highly elevated apoB levels. Thirdly, and maybe most importantly, atherogenesis, and the prevention of ASCVD, is only to a certain extent mediated by apoB levels. ApoB-containing particles are necessary but are not in and of themselves sufficient for the disease process to occur. The pharmacological tools in our proposal and the strength of evidence in support of lowering apoB have gained center stage when it comes to the prevention of MACE and mortality. Still, other risk factors and markers that, in combination with apoB particles, are sufficient for atherogenesis, should not be overlooked.
The efficacy of statin treatment in preventing major adverse cardiac events and reducing all-cause mortality can’t solely be explained by the achieved reductions in apoB. A number of papers have pointed out the pleiotropic effects of statins in reducing inflammatory markers, raising awareness of the importance of managing inflammation in the prevention of atherogenesis and ASCVD. Epidemiological studies, as well, are pointing in the direction of inflammation being of major importance with regard to risk assessment:
With respect to the primary endpoint of combined MI and ischemic stroke, in unadjusted analyses, GlycA and hsCRP were both directly associated with the primary endpoint. Both GlycA and hsCRP remained independently associated with the primary endpoint in adjusted analyses.3

In an expert interview by Medscape4, Paul M. Ridker describes the emergent utility of high-sensitivity C-reactive protein (hsCRP) in risk assessment:
It is overwhelmingly clear that CRP, or more specifically, high-sensitivity C-reactive protein (hsCRP), is an important marker of risk that adds prognostic information at all levels of low-density lipoprotein (LDL)-cholesterol, at all levels of the metabolic syndrome, and at all levels of the Framingham Risk Score. hsCRP levels of < 1, 1-3, and > 3 mg/L correspond with lower, moderate, and higher vascular risk across wide groups of patients.
The association between hsCRP and adverse cardiovascular events raises many questions. Does hsCRP reduction correlate with lipid reductions? Is hsCRP a proxy for some underlying pathological process or in and of itself a causal actor? What’s the relationship between other known risk markers and hsCRP? The list goes on…
As Ridker goes on to describe, hsCRP does not correlate with LDL-c and is predictive of adverse cardiac events independently of cholesterol levels:
… physician cannot predict which patients are going to get a CRP reduction on the basis of LDL-cholesterol reduction. They have to measure both LDL-cholesterol and hsCRP. I would argue that the new data strongly suggest that we need to measure and manage CRP in the same way that we currently measure and manage LDL-cholesterol, if we are going to achieve the greatest benefits for our patients.
Ridker et al. have demonstrated this fairly consistently by running subanalyses of randomized, place-controlled trials done on both statins and EPA. In Measurement of C-Reactive Protein for the Targeting of Statin Therapy in the Primary Prevention of Acute Coronary Events5, Ridker et al. "measured the level of C-reactive protein both at baseline and after one year of follow-up among 5742 of the 6605 participants enrolled in a randomized, double-blind, placebo-controlled trial of lovastatin in the primary prevention of acute coronary events in persons with average levels of total cholesterol and below-average levels of high-density lipoprotein (HDL) cholesterol."
Our data provided minimal evidence of an association between base-line C-reactive protein levels and base-line lipid levels; the Spearman correlation coefficients for the relations between C-reactive protein levels and total, LDL, and HDL cholesterol and triglyceride levels and the ratio of total to HDL cholesterol were 0.069, 0.012, –0.058, 0.129, and 0.092, respectively. Thus, less than 2 percent of the variance in base-line C-reactive protein levels was determined by lipid factors.
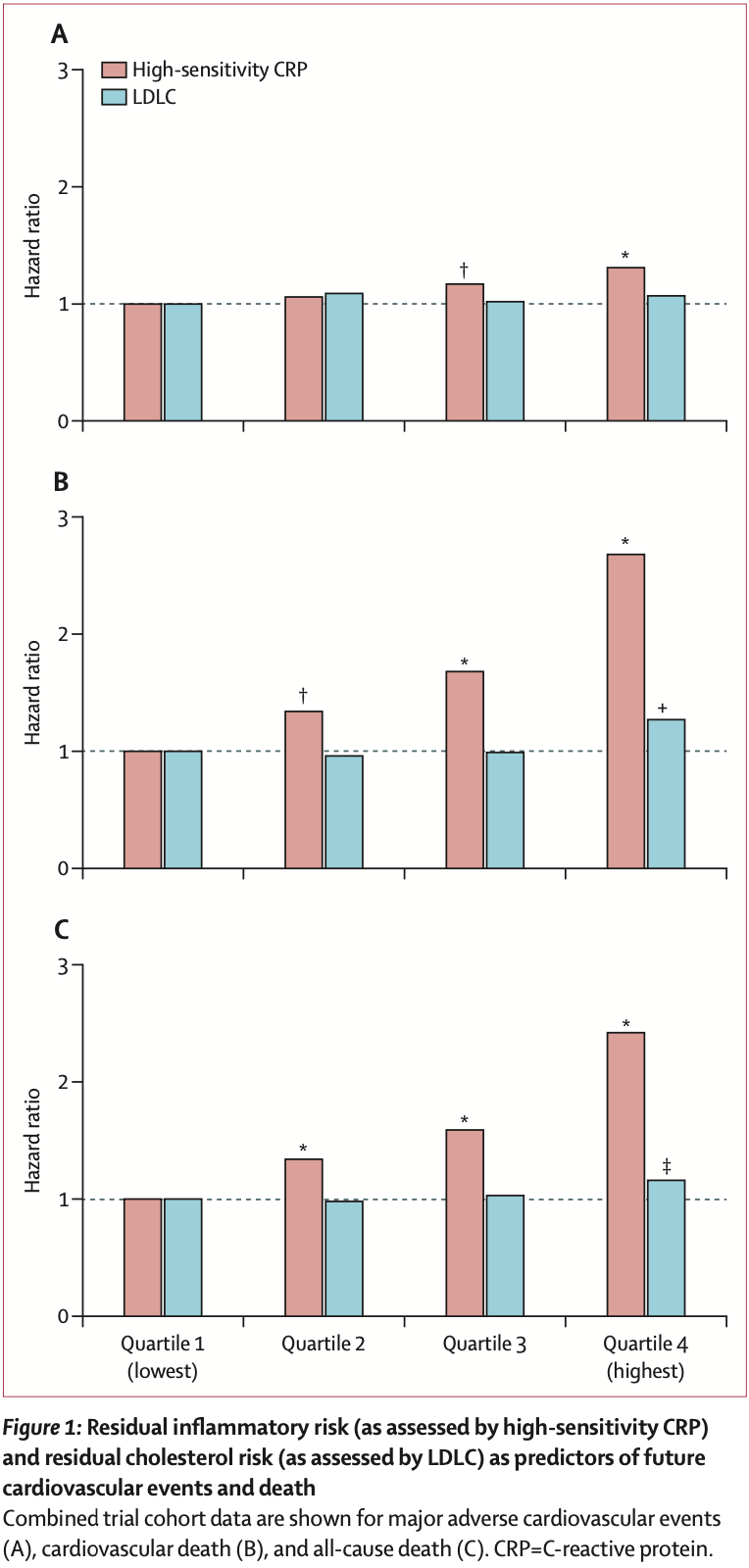
In Inflammation and cholesterol as predictors of cardiovascular events among patients receiving statin therapy: a collaborative analysis of three randomized trials6, Ridker et al. ran an analysis of PROMINENT, REDUCE-IT, and STRENGHT7, where they 1) assessed the "hazard ratios (HRs), 95% CIs, and p values for the occurrence of incident major adverse cardiovascular events, cardiovascular mortality, and all-cause mortality by quartiles of baseline high-sensitivity CRP and LDLC, by use of trial-specific quartile cutpoints to minimize any bias between trials.", 2) ran a fixed-effects meta-analysis of the three trials, and 3) “each trial analysis group was additionally asked on an a priori basis to compute risk HR estimates across four predefined residual risk groups that have consistently been used in previous clinical studies.”8
Again, the findings by Ridker et al. support the predictive role of hsCRP, independent of LDL-c:
In all three trials, baseline high-sensitivity CRP was a significant predictor of incident major adverse cardiovascular events, cardiovascular death, and all-cause death, with no significant heterogeneity for any of the three endpoints.
In all three trials, the relationships of baseline LDLC to subsequent cardiovascular event risk were less robust than for high-sensitivity CRP, again with no significant heterogeneity between trials
Both of the analyses mentioned above did also demonstrate that when it comes to hsCRP “lower is better” with regard to MACE, and even CVD and all-cause mortality:
Overall, the rates of coronary events increased with the base-line levels of C-reactive protein, so that the relative risks of coronary events in participants assigned to the placebo group as compared with those in the lovastatin group were 1.0, 1.2, 1.3, and 1.7 for the lowest to highest quartile of base-line levels of C-reactive protein. In unadjusted analyses, the risk of acute coronary events increased by 21 percent with each increasing quartile of base-line C-reactive protein levels (95 percent confidence interval, 4 to 41 percent).
For major adverse cardiovascular events, the pooled HRs for the lowest to highest quartiles of high-sensitivity CRP were 1 (ref), 1.06 (95% CI 0.97–1.16), 1.17 (1.07–1.28), and 1.31 (1.20–1.43). For cardiovascular death, the pooled HRs for the lowest to highest quartiles of high-sensitivity CRP were 1 (ref), 1.34 (95% CI 1.09–1.64), 1.68 (1.38–2.04), and 2.68 (2.22–3.23). For all- cause death, the pooled HRs for the lowest to highest quartiles of high-sensitivity CRP were 1 (ref ), 1.34 (95% CI 1.16–1.55), 1.59 (1.38–1.83), and 2.42 (2.12–2.77).
To further harp on the findings above, let's mention a few more studies. Nissen et al. did demonstrate that atheroma progression is correlated with CRP levels.9 The researchers performed intravascular ultrasonography on participants who had stenosis of at least 20 percent on angiography and who had undergone a statin-free washout period of 4 to 10 weeks. After this, the participants underwent randomization to receive either 40 mg of pravastatin or 80 mg of atorvastatin daily and were followed up for 18 months.
In contrast to Ridker et al., Nissen et al. did demonstrate a correlation between CRP and LDL-c:
There was a weak but significant correlation between the percent reductions in LDL cholesterol and in CRP levels only for the study group as a whole — not for the pravastatin group alone or the atorvastatin group alone. Changes in other atherogenic lipoproteins, such as apo B-100 and non-HDL cholesterol, had similarly weak correlations with the reduction in CRP levels in the regression analysis.
These findings are not unexpected though. Since both groups did undergo a washout period before starting statin therapy, we can expect the lowering of both LDL-c and CRP upon initiation of treatment.
As could be expected by now, the lowering of both CRP and LDL-c correlated with slower atheroma progression. Moreover, the largest CRP reduction did correlate with atheroma regression:
…greater reductions in LDL cholesterol levels were associated with slower rates of progression on intravascular ultrasonography. [figure 2] shows this same relationship for the reduction in CRP levels. Patients with the largest reductions in CRP levels had regression of atheroma, as evidenced by progression rates of less than zero.
Now, since the LDL-c and CRP levels did correlate, and since the nature of this study cannot infer causality for either of the markers measured, we’d like to tease out the relative contributions of LDL-c and CRP lowering to atheroma development.
…correlations between the rates of progression on ultrasonography and the percent change in non-HDL cholesterol, LDL cholesterol, and CRP levels remained significant on multivariate analysis but were weaker than those obtained by univariate analyses.

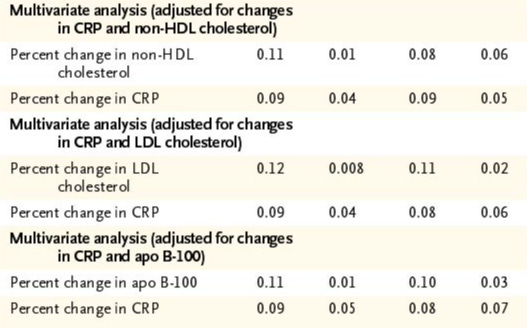
A closer view of the numbers reveals that reductions in both lipid markers and CRP did independently correlate with atheroma progression, although with more uncertainty than we might be comfortable with.

This being said, based on table 4 above, it seems like CRP alone did correlate with rates of progression quite substantially, which led Nissen et al. to conclude:
Patients with reductions in the levels of both LDL cholesterol and CRP that were greater than the median reduction had significantly lower progression rates than patients in whom the reductions were less than the median decrease. These data provide evidence that the reduction in CRP levels plays an independent role in the beneficial effects of statins on the progression of coronary atherosclerosis.
Ridker, in the Medscape interview, does also emphasize the importance of these findings, referring to further analyses done on the same data:
Those who only got the LDL-cholesterol down had some slowing of the progression; those who only got the CRP down actually got regression; and those who got both the cholesterol and the CRP down got the greatest regression.
…the investigators were able to measure the change in CRP from beginning to end, and they also observed, as we did, that there is almost no relationship between the change in CRP and the change in LDL-cholesterol. Once again, therefore, the conclusion that must be drawn is that if you want your patient to get the greatest benefit from these drugs, that is, actual regression and the lowest event rates, it is not enough to lower their LDL-cholesterol; they need to lower their CRP as well.
So these 2 observations together produce a very powerful shift in the entire paradigm, which is simply that from a clinician's point of view: In order to optimize the benefit for the patient, it is no longer enough to lower the LDL-cholesterol; we need to measure and monitor the CRP and treat that the same way we treat cholesterol.
Masson et al., in their analysis of the A to Z trial,10 did evaluate the utility of hsCRP in predicting future outcomes among patients with non–ST-elevation and ST-elevation acute coronary syndrome.11 hsCRP was measured 30 days after the initial onset of acute coronary syndrome and at the subsequent month 4 and month 8 visits. The participants were randomized at the onset of ACS to receive 40 mg/d of simvastatin for one month followed by 80 mg/d thereafter, or placebo for 4 months and 20 mg/d simvastatin thereafter.
This analysis by Masson et al. includes an iconic Kaplan-Meier curve that describes the relationship between achieved hsCRP and LDL-c levels and the cumulative probability of either death or myocardial infarction:

Again, in support of the narrative painted by the previous finding by both Ridker et al. and Nissen et al., Masson et al. go on to conclude:
After adjusting for age, gender, diabetes mellitus, smoking, history of known cardiovascular disease, index event, achieved LDL, use of estrogen hormonal replacement therapy, treatment with ACE inhibitors, and the study treatment allocation, patients with a level of hsCRP >3 mg/L had a >3-fold-higher risk of death (adjusted HR, 3.7; 95% CI, 1.9 to 7.2) compared with patients with a level of hsCRP <1 mg/L.
Furthermore,
…the relation between the achieved level of LDL cholesterol and subsequent outcome was less robust than that observed for hsCRP. Specifically, patients with LDL <70 mg/dL at 30 days were at similar risk for death and major cardiovascular events as those with LDL above this cut-point. Assessed at 4 months, the risk of subsequent major cardiovascular events tended to be lower in patients with LDL <70 mg/dL.
Stratification by the achieved level of hsCRP and LDL at 4 months revealed a pattern consistent with that observed in our prior study, with highest risk in those who failed to achieve either hsCRP <2 mg/dL or LDL <70 mg/dL, intermediate risk in those who achieve one but not both goals and lowest risk in those who achieved these potential dual goals of therapy.
There have been hints of the following already, but in order for hsCRP, or other inflammatory markers to be pragmatically interesting, they’ll need to be modifiable, and their modification should impact outcomes. The studies mentioned above do clearly point in the direction of this being the case with hsCRP, but it’s unclear how hsCRP, or inflammation overall, fits into the framework of being necessary and/or sufficient, or neither.
All the usual “lifestyle factors” play their part in one’s overall inflammatory status, but it’s evident that some residual inflammation might be present even when everything is seemingly optimized. Also, we need to recognize that everything can’t always be viewed through optimizations and concessions are to be made, in which case we’d like to have tools for risk mitigation.
In the JUPITER trial12, Ridker et al. investigated the impact of lowering hsCRP in “healthy” participants (41.4% had metabolic syndrome, but had no history of cardiovascular events) with median levels of LDL-c and hsCRP of 108 mg/dL and 4.2 mg/dL, respectively. Median BP was 134/80 mmHg and HbA1C 5.7%.
The treatment group who were assigned to 20mg of rosuvastatin daily did experience an approx. 50% reduction in both LDL-c (55 mg/dL) and hsCRP (2.2 mg/L) at 12 months follow-up.
JUPITER was terminated early due to the overwhelming superiority of rosuvastatin over placebo:
At the time of study termination (median follow-up, 1.9 years; maximal follow-up, 5.0 years), 142 first major cardiovascular events had occurred in the rosuvastatin group, as compared with 251 in the placebo group. The rates of the primary end point were 0.77 and 1.36 per 100 person-years of follow-up in the rosuvastatin and placebo groups, respectively.
On the basis of Kaplan–Meier estimates, the number of patients who would need to be treated with rosuvastatin for 2 years to prevent the occurrence of one primary end point is 95, and the number needed to treat for 4 years is 31. If 4-year risks are projected over an average 5-year treatment period, as has been commonly done in previous statin trials according to the method of Altman and Andersen, the number needed to treat to prevent the occurrence of one primary end point is 25.

Subanalyses of the results revealed that the benefits of rosuvastatin extended beyond the subpopulations with metabolic syndrome or any other risk factors:
For the primary end point, there was no evidence of heterogeneity in the results for any subgroup evaluated.
Groups typically assumed to be at very low risk also benefited.
High-sensitivity C-reactive protein did once again emerge as a target of treatment whose reduction, independently of lipid levels, reduced risk:
For participants who had elevated levels of high-sensitivity C-reactive protein but who were nonsmokers, were not overweight (BMI ≤ 25), did not have the metabolic syndrome, had a calculated Framingham risk score of 10% or less, or had an LDL cholesterol level of 100 mg per deciliter (2.6 mmol per liter) or lower, the observed relative reductions in the hazard ratio associated with rosuvastatin for the primary end point were similar to those in higher-risk groups.
For subjects with elevated high-sensitivity C-reactive protein levels but no other major risk factor other than increased age, the benefit of rosuvastatin was similar to that for higher-risk subjects (hazard ratio, 0.63; 95% CI, 0.44 to 0.92; P=0.01)

This far, based on the subanalyses and data from clinical trials, some of which are discussed above, we can be quite confident that reducing hsCRP will reduce atherogenesis and the risk of MACE. Confidence, however, is relative, and shouldn’t be confused with certainty.
Trials done on anti-inflammatory agents, such as colchicine13 and canakinumab14, have also demonstrated benefits from reducing inflammatory burden on cardiovascular disease:
For the key secondary cardiovascular end point, the incidence rate was 5.13 events per 100 person-years in the placebo group, 4.56 events per 100 person-years in the group that received the 50-mg dose of canakinumab, 4.29 events per 100 person-years in the 150-mg group, and 4.25 events per 100 person-years in the 300-mg group.
…colchicine significantly reduced hsCRP (-5,50 vs. -1,6 pg/dL) comapred to controls…
Colchicine treatment was associated with lower risk of MACE, recurrent MI, stroke and hospitalization due to CV causes compared to the control group.
These treatments are of course not without their issues but do demonstrate the relationship between reductions in inflammation and the decreased risk of adverse cardiovascular outcomes, beyond the ones observed with statins.
Lastly, to further strengthen our confidence in these findings, mendelian randomization studies have demonstrated a relationship between the IL-6 pathway upstream of hsCRP and cardiovascular disease.15 Findings by Cupido et al. demonstrated that:
The Mendelian randomization analysis of IL6-receptor [and IL6 levels themselves] suggested that 1 mg/L lower CRP levels due to the IL6-receptor instrument are associated with a lower risk for coronary artery disease, any stroke and any ischaemic stroke, atrial fibrillation and rheumatoid arthritis.
The IL6 level or -receptor instruments did not associate with any changes in meaningful changes to basic lipid measurements, which again might indicate the independent nature by which inflammation contributes to cardiovascular risk.
Referring to the framework laid out earlier, there seems to be quite a substantial amount of evidence indicating that markers of inflammation do offer us additional benefits beyond traditional risk markers, such as lipids and blood pressure. Again, we can never attain certainty, but in light of the current evidence it would not be ill-informed to monitor inflammation and adjust interventions accordingly when assessing cardiovascular and -metabolic risk, especially in patients with stubborn lipids or other major risk factors and markers.
…participants with neither residual inflammatory risk nor residual cholesterol risk (high-sensitivity CRP <2 mg/L and LDLC <70 mg/dL; the reference group), residual cholesterol risk only (high-sensitivity CRP <2 mg/L and LDLC ≥70 mg/dL), residual inflammatory risk only (high-sensitivity CRP ≥2 mg/L and LDLC <70 mg/dL), and both residual cholesterol risk and residual inflammatory risk (high-sensitivity CRP ≥2 mg/L and LDLC ≥70 mg/dL).